
Define how transcription factor and epigenetic determinants of cell identity are disrupted in pancreas disease
Insults such as inflammation, obesity, and an oncogenic KRAS mutation destabilize cell identity in the pancreas and collude in the formation of pancreatic cancer. How do these insults disrupt the transcription factor networks and epigenetic landscapes that establish cell identity? Can these disruptions be targeted to prevent formation of pancreatic cancer? In the Nissim lab, we are pioneering cutting-edge technologies never before applied to the pancreas in order to answer these questions.
Discover and characterize novel cancer-causing genes in unsolved families
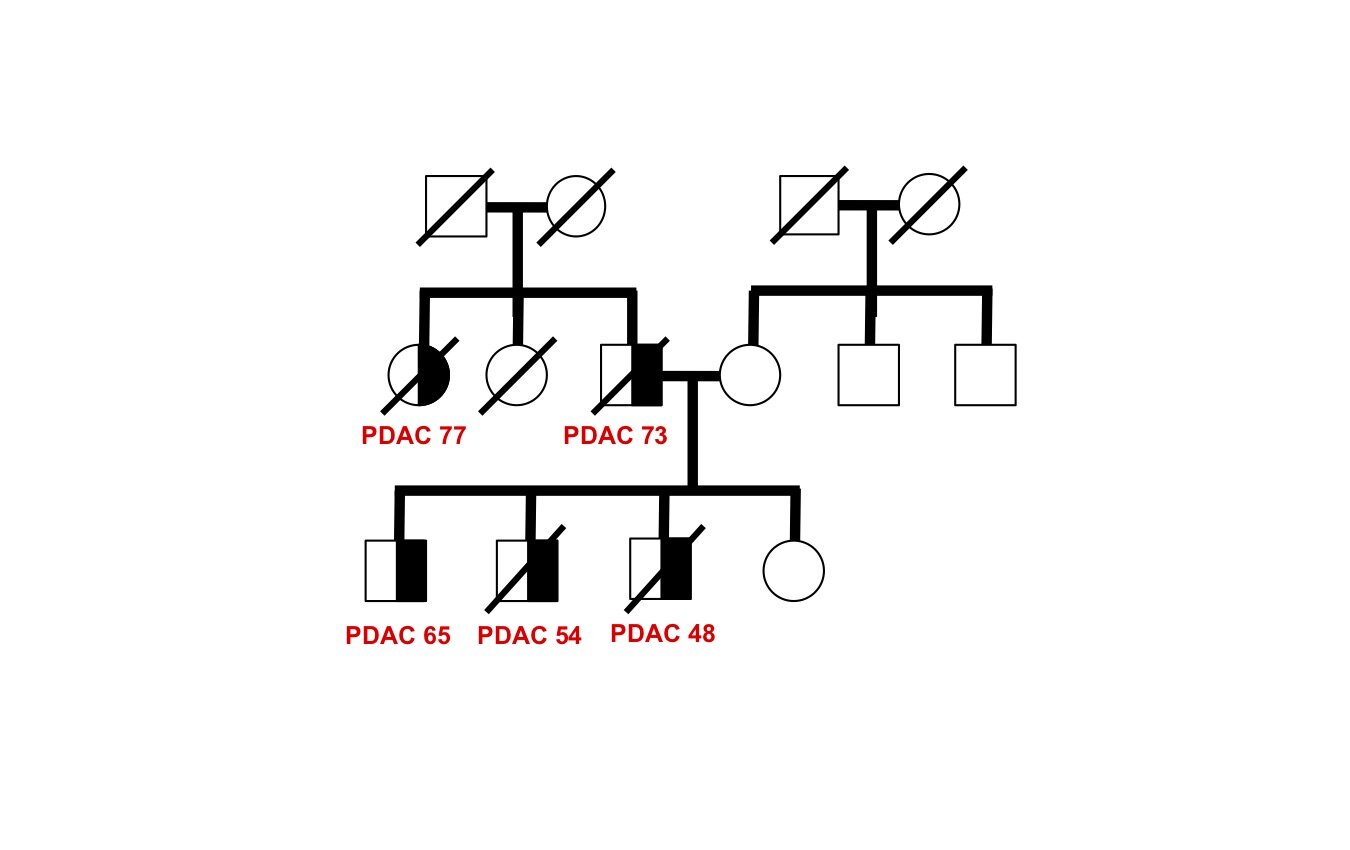
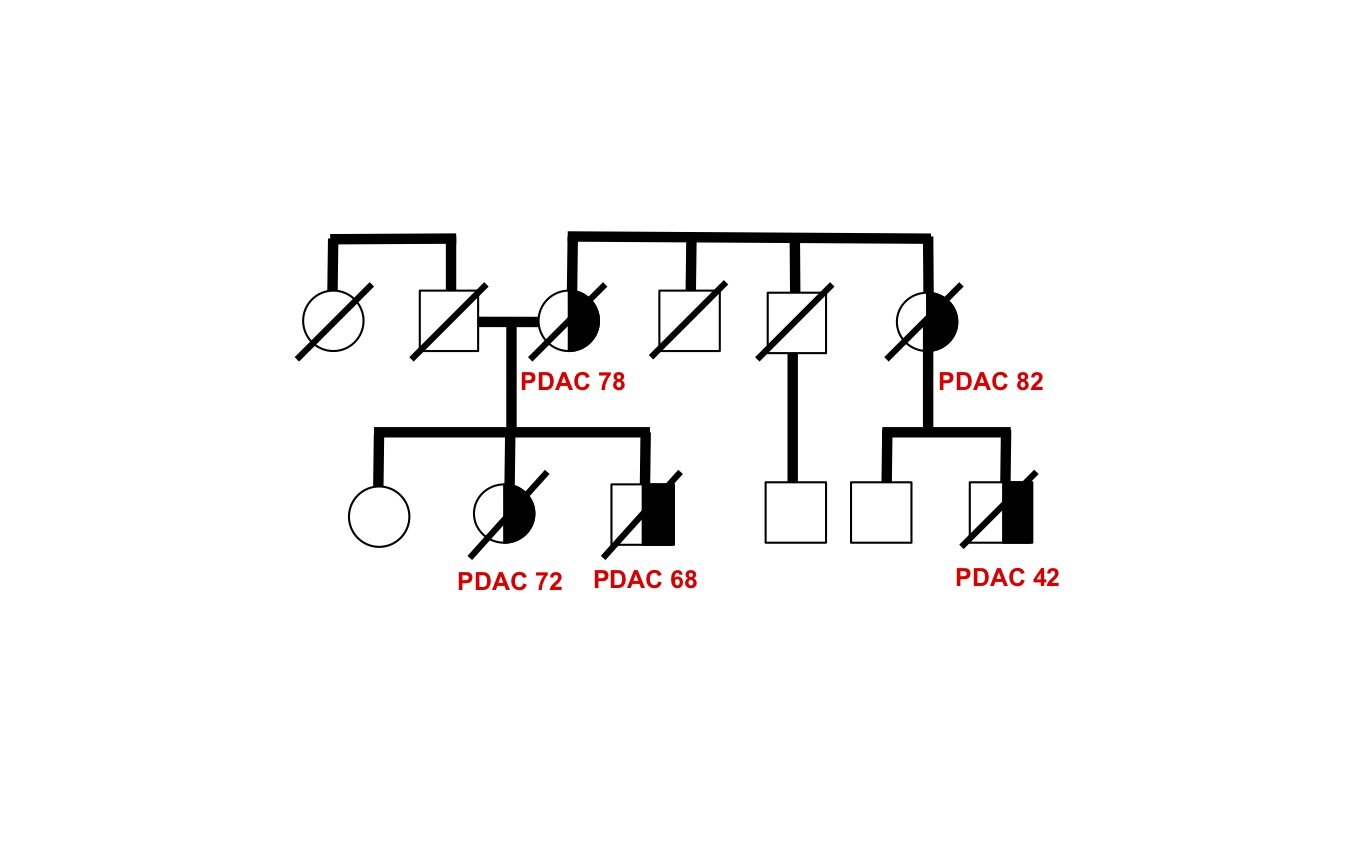
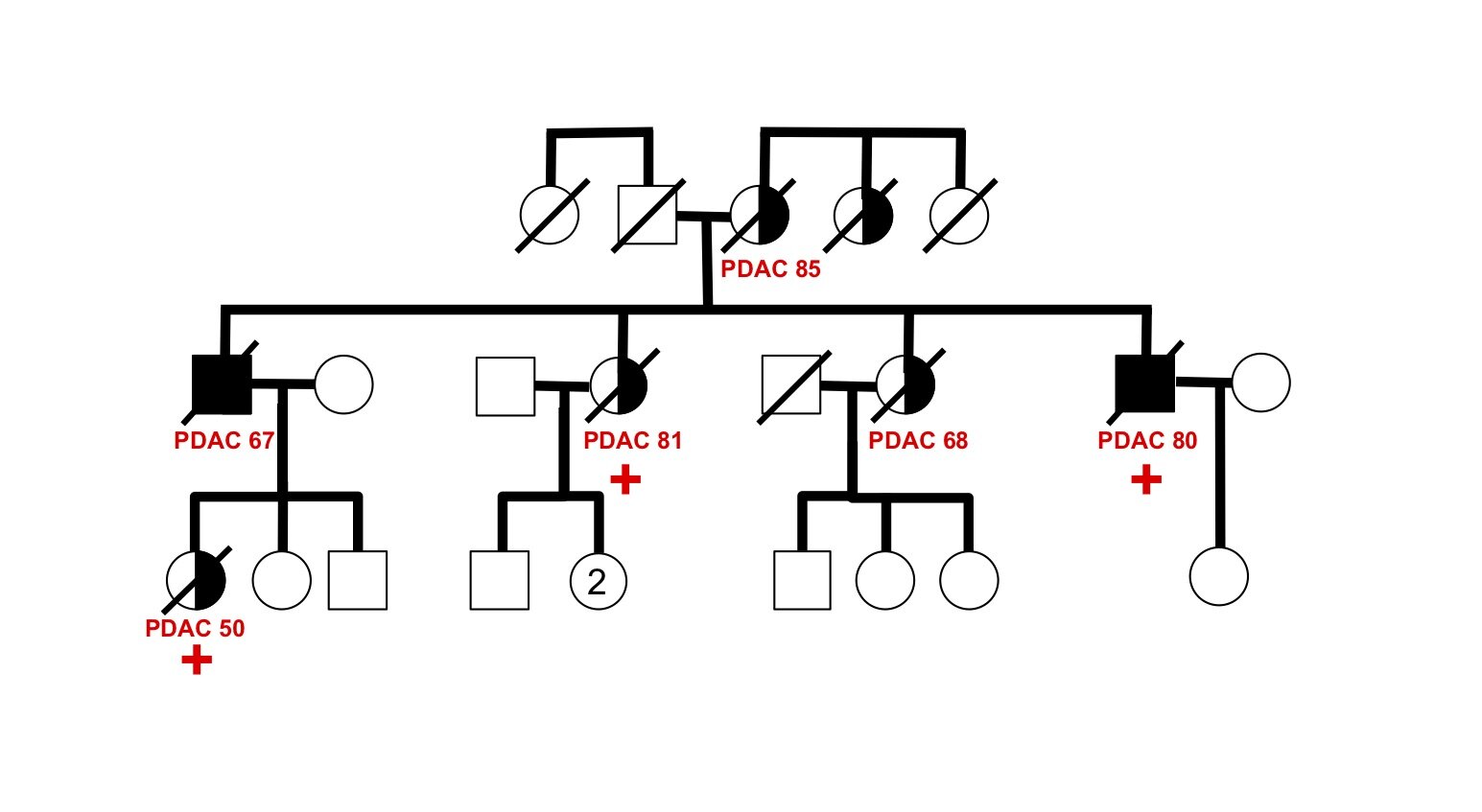
The genetic basis of most hereditary pancreatic cancer has not been determined, yet understanding these cancer-causing genes gives us precious clues into pathogenesis and new strategies for treatment and prevention. Each affected family has the potential to contribute fundamental new insights in cancer biology — there are many “unsolved” hereditary pancreatic cancer families awaiting future study.
As a proof-of-principle, we performed whole genome sequence analysis on a family with 5 cases of pancreatic cancer and identified a rare mutation in a gene RABL3 not previously associated with cancer. By recapitulating this mutation in large zebrafish population studies, we validated its role in promoting cancer. We further discovered that the mutation alters a key step in the intracellular trafficking of KRAS, one of the most common drivers in cancer. We are now exploring the therapeutic potential of targeting this node in RAS-driven cancers and RASopathy syndromes. This work (Nissim et al., Nature Genetics 2019) demonstrates that each hereditary pancreatic cancer family has the potential to uncover fundamental insights into cancer biology with broad translational implications.

Develop new small molecule therapies to target RAS-driven cancers

Building on our discovery that RABL3 regulates prenylation and trafficking of KRAS, we are exploring the therapeutic potential of disrupting RABL3 activity to target RAS-driven cancers. Projects related to the RAS pathway include developing a biochemical screen for small molecules that disrupt RABL3-RAP1GDS1 interactions, as well as an in vivo (phenotypic) screen for pharmacological agents that rescue RAS over-activation in larval zebrafish. Positive hits from these screens will lead to pre-clinical studies in zebrafish models of RASopathy and cancer.
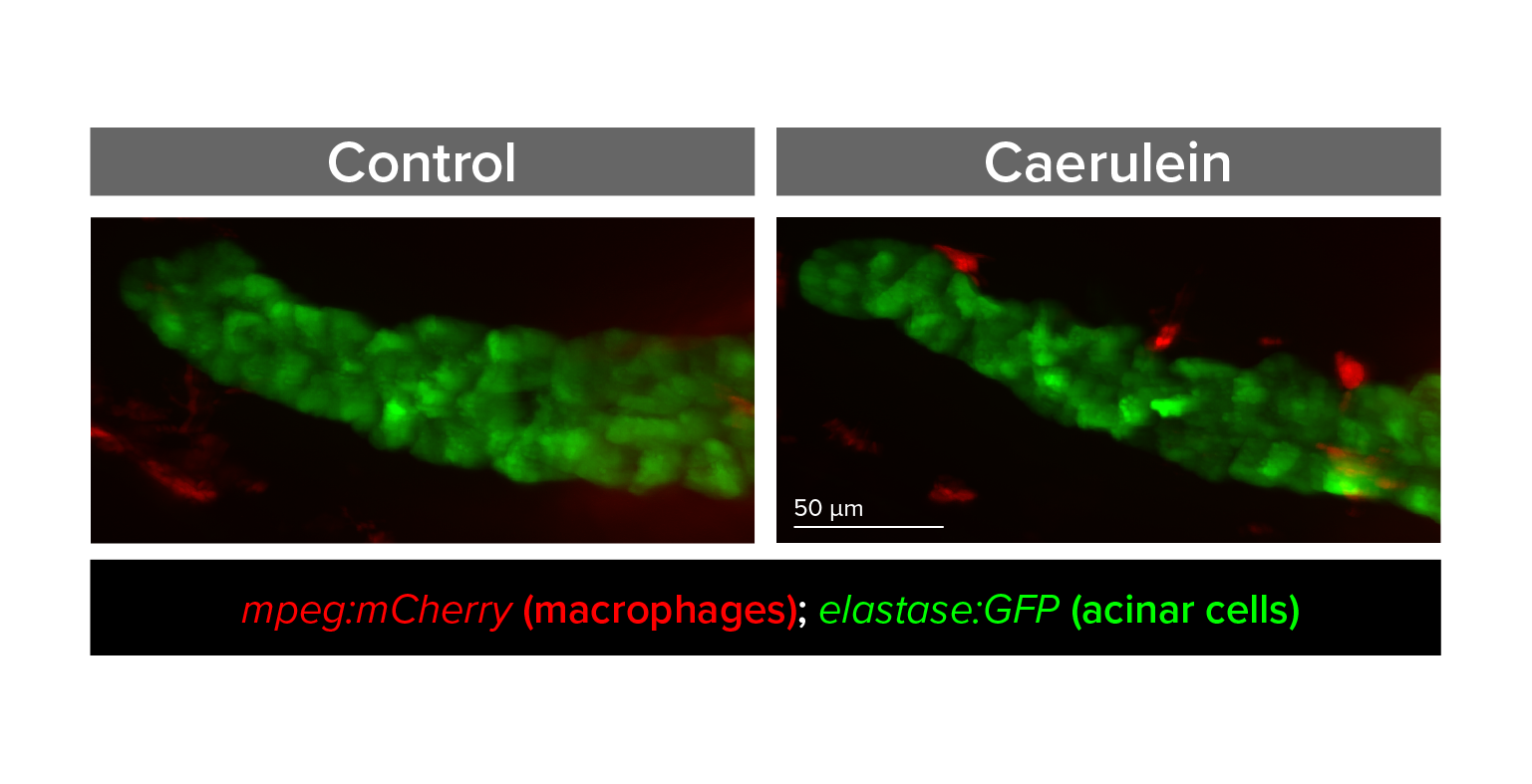
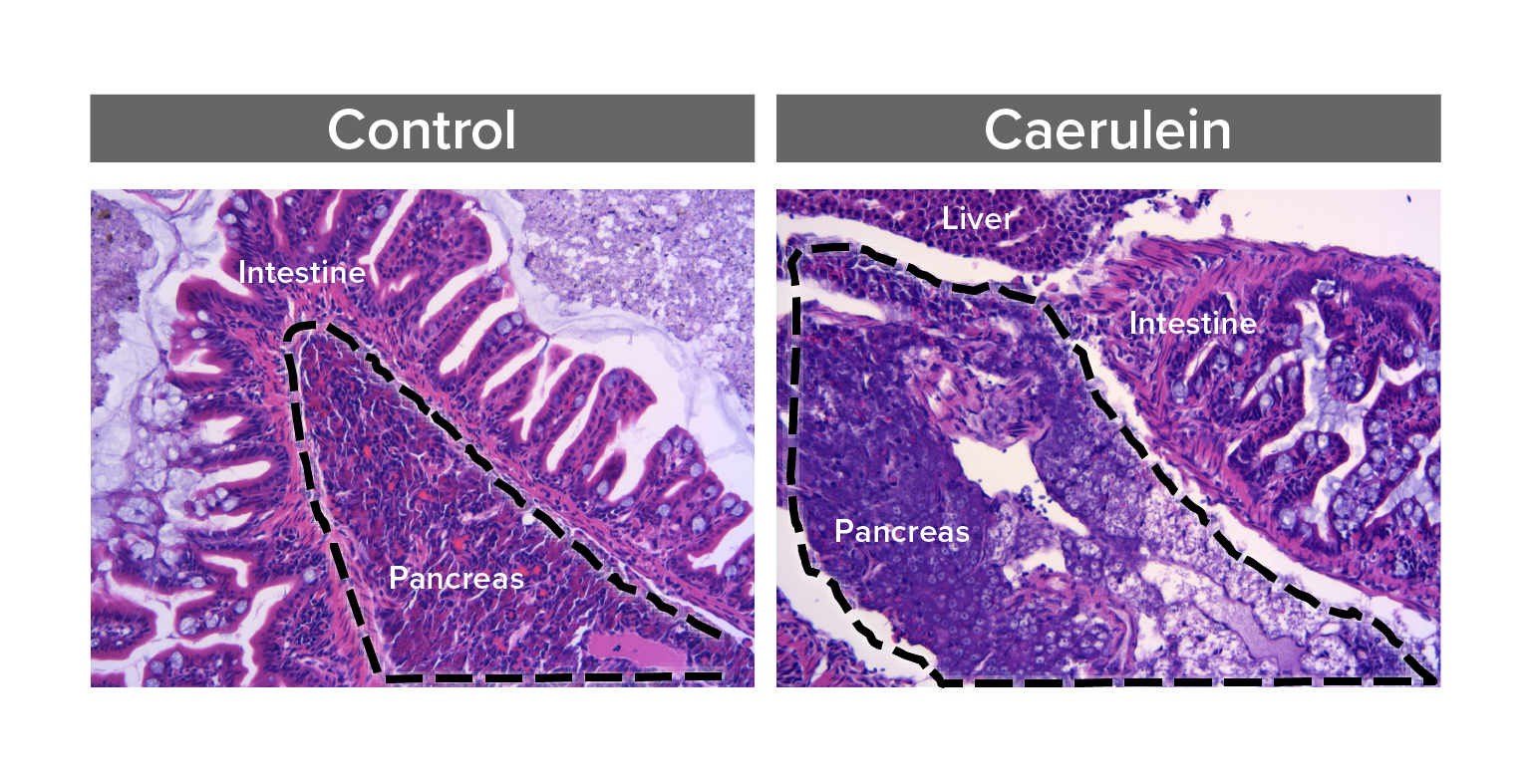
Study pathways that regulate pancreas inflammation and regeneration
Acute pancreatitis is one of the top 5 most frequent causes of U.S. hospital admission, yet pathophysiology is poorly understood and treatment options are limited. We are developing zebrafish models that for the first time would allow in vivo, real-time, and longitudinal study of cellular processes, inflammation, and signaling pathways involved in pancreatitis.
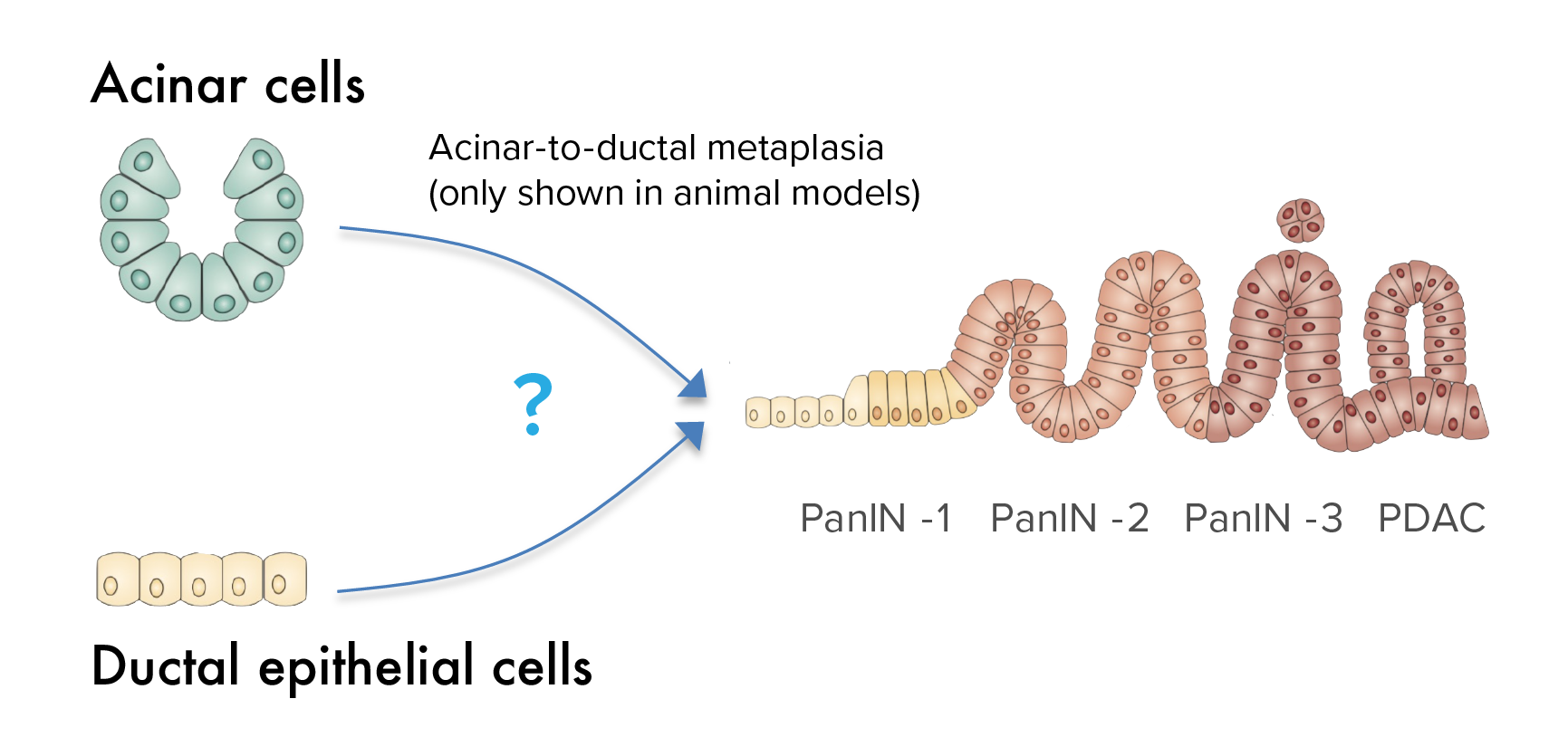
Determine the cell of origin of pancreatic cancer
Acinar and ductal epithelial cells are the most abundant cell populations in the exocrine pancreas, and there is a longstanding debate about which cell leads to pancreatic cancer. While pancreatic cancer most often exhibits histologic features of a ductal origin, animal studies suggest that pancreatic cancer arises from acinar but not ductal cells, suggesting that a “metaplasia” process is necessary for cancer initiation. This has never been shown in human tissue. This question has important implications for understanding the processes by which a normal cell transforms into a cancer cell. We are undertaking single cell approaches to definitively determine the clonal origin in human samples.
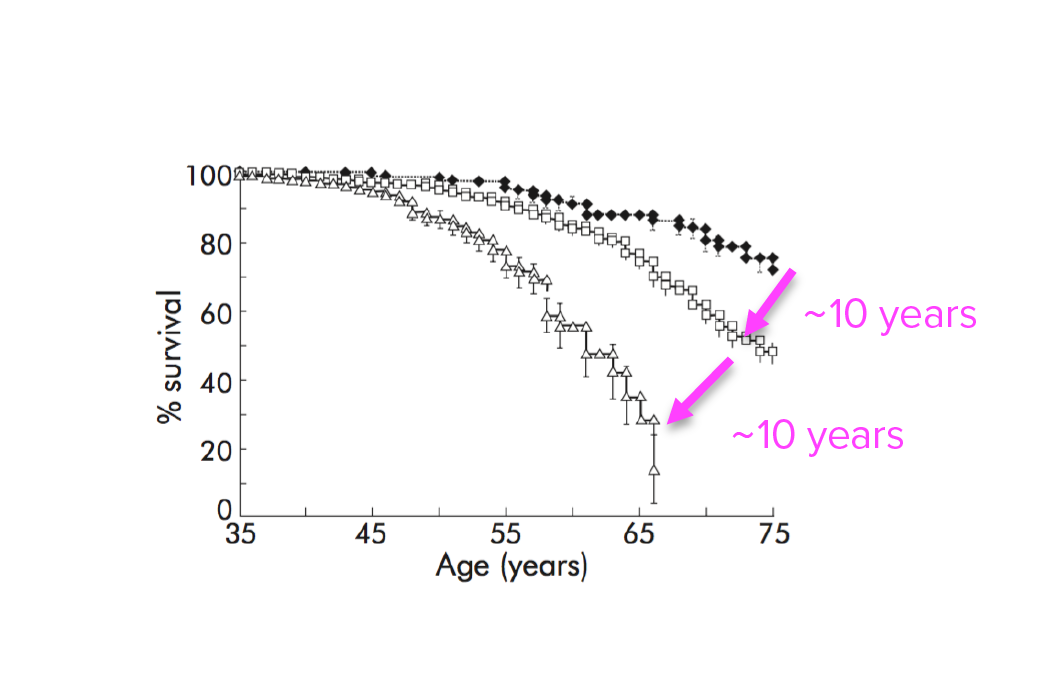
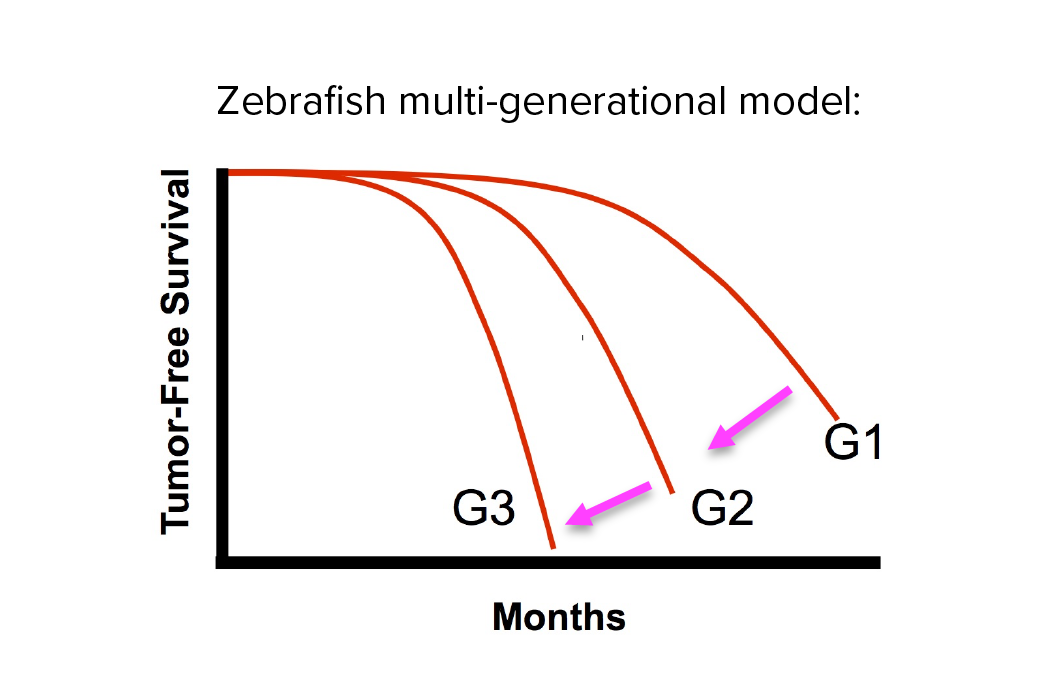
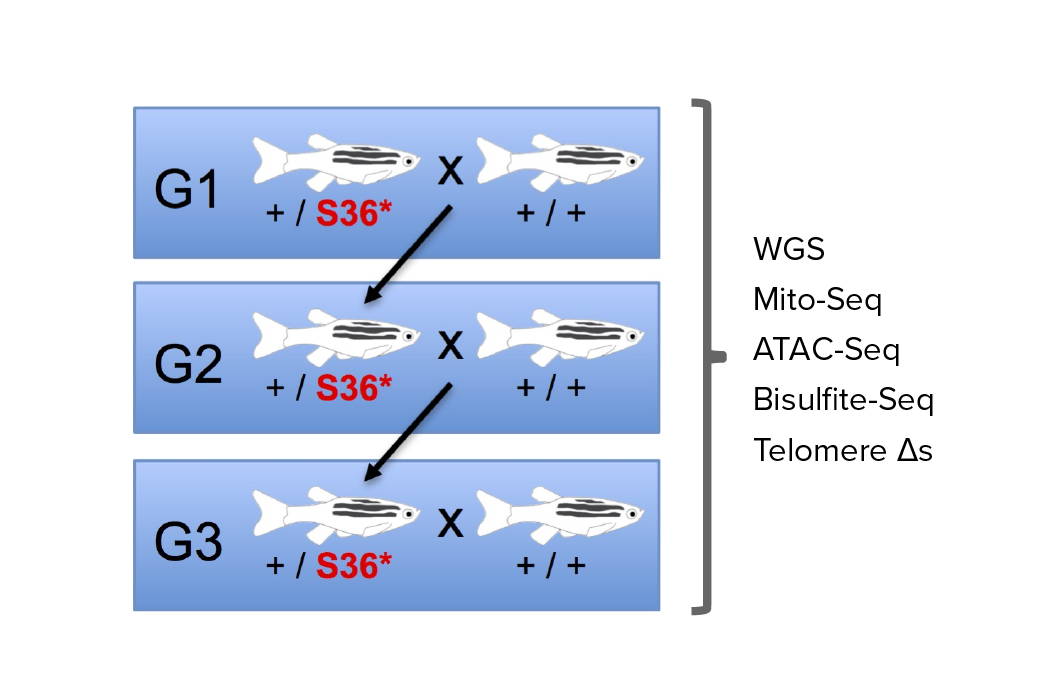
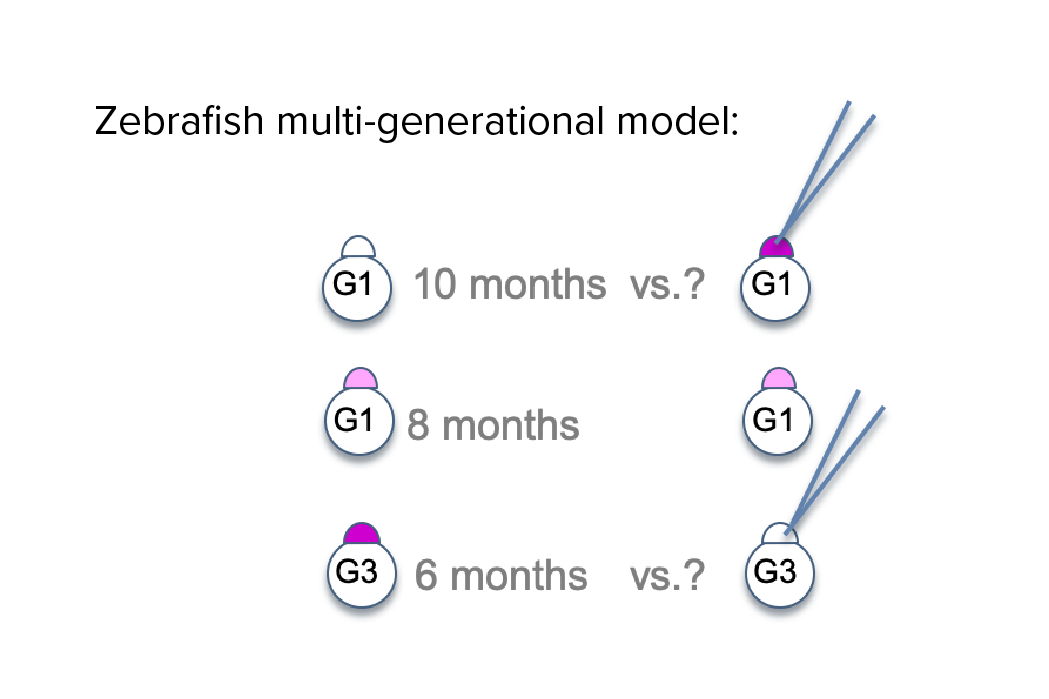
Investigating transgenerational anticipation in hereditary pancreatic cancer families
In families with hereditary pancreatic cancer, it is a longstanding yet unexplained observation to see progressively younger ages of onset in each generation. This biological phenomenon can be modeled with multi-generation zebrafish families, enabling a mechanism to be identified and tested.